u2.08.06 Notice d’utilisation du parallélisme#
Résumé:
Toute simulation Code_Aster peut bénéficier des gains de performance que procure le parallélisme (HPC [1]_ en anglais). Ces gains peuvent être de deux ordres: sur le temps de calcul et sur l’espace RAM/disque requis (par cœur alloué). Bien compris et bien utilisé, l’impact du HPC sur les études peut être majeur en termes de:
Faisabilité: débloquer une situation critique (modèle trop fin, simulation trop lente…);
Productivité: ses accélérations jusqu’à X10 voire X100 [2]_
contribuent à fluidifier et à sécuriser la conduite des simulations;
Prédictivité: permettre des modèles plus fins, des simulations plus complexes et mieux validées (recalage, sensibilité, paramétrique…);
Prospective: se permettre des études complètement différentes.
Code_Aster propose différentes stratégies parallèles pour s’adapter à l’étude et à la plate-forme de calcul. Certaines sont plutôt axées sur des aspects informatiques (distribution de calculs complets ou de calculs modaux/MISS3D indépendants, construction de systèmes linéaires, opérations basiques d’algèbre linéaire), d’autres sont plus algorithmiques (solveurs linéaires HPC MUMPS et PETSc).
Ce document décrit brièvement l’organisation du parallélisme dans le code. Puis il rappelle quelques fondamentaux afin d’aider l’utilisateur à tirer parti de ces stratégies parallèles. On détaille ensuite leurs mises en œuvre, leurs périmètres d’utilisation et leurs gains potentiels. Les chaînages/cumuls de différentes stratégies (souvent naturels et paramétrés par défaut) sont aussi abordés.
L’utilisateur pressé peut d’emblée se reporter au chapitre 2 («Le parallélisme en quelques clics !»). Il résume le mode opératoire pour mettre en œuvre la stratégie parallèle préconisée par défaut.
Remarque:
Pour utiliser Code_Aster en parallèle, (au moins) trois cas de figures peuvent se présenter:
On a accès à la machine centralisée Aster et on souhaite utiliser l’interface d’accès Astk,
On effectue des calculs sur un cluster ou sur une machine multi-c œ urs avec Astk,
Idem que le cas précédent mais sans Astk.
Généralités#
Toute simulation Code_Asterpeut bénéficier des gains de performance que procure le parallélisme. Du moment qu’il effectue des calculs élémentaires/assemblages, des résolutions de systèmes linéaires, de gros calculs modaux ou des simulations indépendantes/similaires. Les gains peuvent être de deux ordres : sur le temps de calcul et sur l’espace RAM/disque requis (par processus MPI).
Comme la plupart des codes généralistes en mécanique des structures, Code_Aster propose différentes stratégies pour s’adapter à l’étude et à la plate-forme de calcul. Une fois paramétrées, la plupart s’enchaînent et se couplent de manière automatique. Les paragraphes suivant en font le synoptique.
Parallélismes informatiques#
1a/Lancement de rampes de calculs indépendants/similaires (calculs paramétriques, tests…).
Outil: scriptage shell.
Gain : en temps elapsed.
Lancement: standard via Astk[U2.08.07].
Chaînage : aucun.
Cumul : possibles avec les autres schémas parallèles mais uniquement en usage avancé (surcharge de sources).
1b/Distribution des calculs élémentaires et des assemblages matriciels et vectoriels dans les pré/post-traitements et dans les constructions de systèmes linéaires. Parallélisme en mémoire distribuée.
Outil: langage MPI.
Gain : en temps elapsed, voire en pic mémoire RAM avec MUMPS+MATR_DISTRIBUEE.
Lancement: standard via Astk (mpi_nbcpu/mpi_nbnœud).
Chaînage : utile avec 2b ou 2c; possible mais peu utile avec 1c ou 1d seuls; possible mais inutile avec 2a.
Cumul : aucun.
1c/Distribution des calculs d’algèbre linéairebasiques (sous-étapes de MUMPS, bibliothèque BLAS) *.* Parallélisme en mémoire partagée activé uniquement avec le package MUMPS (cf. 2b) .
Outil: langage OpenMP.
Gain : en temps elapsed (mais augmentation du temps CPU).
Lancement: standard via Astk (ncpus).
Chaînage : possible mais peu utile avec 1b seul.
Cumul : contre-productif avec 2a, utile avec 2b, possible mais peu utile avec 2c ou 1d.
1d/Distribution des calculs modaux (resp. des calibrations modales) dans l’opérateur CALC_MODES (resp. INFO_MODE ). Parallélisme en mémoire distribuée (sans gain mémoire, uniquement accélération).
Outil: langage MPI.
Gain : en temps elapsed (mais augmentation du pic mémoire RAM à contrebalancer par le cumul avec 2b).
Lancement: standard via Astk (mpi_nbcpu/mpi_nbnœud).
Chaînage : possible mais peu utile avec 1b seul.
Cumul : utile avec 2b (voire 2b/1c); possible mais peu utile avec 2a; impossible avec 2c.
1e/Distribution des calculs MISS3D dans l’opérateur CALC_MISS . Parallélisme en mémoire distribuée (si OPTION=FICHIER_TEMPS) et/ou partagée (sans gain mémoire, uniquement accélération).
Outil: langages MPI et OpenMP.
Gain : en temps elapsed.
Lancement: standard via Astk (ncpus, mpi_nbcpu/mpi_nbnœud).
Chaînage : aucun.
Cumul : avec FICHIER_TEMPS deux niveaux de parallélisme possible, MPI x OpenMP.
1f/Autresdistributions de calculs: certaines étapes de l’algorithme de contact-frottement, opérateur REST_SPEC_TEMP. Parallélisme en mémoire distribuée (sans gain mémoire, uniquement accélération).
Outil: langages MPI.
Gain : en temps elapsed.
Lancement: standard via Astk (mpi_nbcpu/mpi_nbnœud).
Cumul : possible mais peu utile avec 1b (OpenMP).
Parallélismes numériques#
2a/Solveur direct MULT_FRONT ; Parallélisme en mémoire partagée.
Outil: langage OpenMP.
Gain : en temps elapsed (mais augmentation du temps CPU).
Lancement: standard via Astk (ncpus).
Chaînage : possible mais peu utile avec 1b, 2b ou 2c.
Cumul : contre-productif avec 1c, possible mais peu utile avec 1d.
2b/ PackageMUMPS (soit en tant que solveur direct, via METHODE=”MUMPS”, soit en tant que préconditionneur de PETSC/GCPC via PRE_COND=”LDLT_SP”).Parallélisme en mémoire distribuée.
Outil: langage MPI.
Gain : en temps elapsed et en pic mémoire RAM.
Lancement: standard via Astk (mpi_nbcpu/mpi_nbnœud).
Chaînage : utile avec 1b ou 2c, possible mais peu inutile avec 2a.
Cumul : utile avec 1c ou 1d.
2c/ Solveur itératifPETSC (avec éventuellement MUMPScomme préconditionneurcf. PRECOND=”LDLT_SP”); Parallélisme en mémoire distribuée.
Outil: langage MPI.
Gain : en temps elapsed et en pic mémoire RAM.
Lancement: standard via Astk (mpi_nbcpu/mpi_nbnœud).
Chaînage : utile avec 1b ou 2b, possible mais inutile avec 2a.
Cumul : possible mais peu utile avec 1c, hors-périmètre avec 1d.
Les schémas parallèles 1b+2b/1c ou 1b+2b+2c sont les plus plébiscités. Ils supportent une utilisation «industrielle» et «grand public». Ces parallélismes généralistes et fiables procurent des gains notables en CPU et en pic RAM par cœur. Leur paramétrisation est simple, leur mise en œuvre facilitée via Astk (cf. §1).
Pour une utilisation standard, l’utilisateur n’a plus à se soucier de la mise en œuvre fine du parallélisme . En renseignant les menus dédiés d’Astk [5] , on fixe le nombre de cœurs requis (pour le MPI et/ou l’OpenMP) ainsi que le nombre de nœuds sur lesquels ils se distribuent.
Parallélisation des systèmes linéaires#
Une fois ces portions de système linéaire construites (schéma parallèle 1b), deux cas de figures se présentent:
soit le traitement suivant est naturellement séquentiel et donc tous les processus MPI doivent avoir accès à l’information globale. Pour ce faire on rassemble ces bouts de systèmes linéaires et donc l’étape suivante ne sera ni accélérée, ni ne verra baisser ses consommations mémoire. Il s’agit le plus souvent d’une fin d’opérateur, d’un post-traitement ou d’un solveur linéaire non parallélisé en MPI (stratégie 1b+2a).
soit le traitement suivant accepte le parallélisme MPI , il s’agit alors principalement des solveurs linéaires HPC MUMPS (1b+2b), PETSC (1b+2c) ou les deux à la fois (1b+2b+2c). Le flot parallèle de données construit en amont leur est alors transmis (après quelques adaptations). Ces packages d’algèbre linéaire réorganisent ensuite, en interne, leurs propres schémas paralléles (avec une vision plus algébrique). On parle alors de schéma parallèle d’ordre plutôt «numérique». Cette combinaison «parallélisme informatique», au niveau de l’assemblage du système linéaire, et, «parallélisme numérique», au niveau de sa résolution, les 2 via MPI, est la combinaison la plus courante.
Remarques:
Notons qu’à l’issue du cycle «construction de système linéaire – résolution de celui-ci», quelque soit le scénario mis en œuvre (solveur linéaire séquentiel ou parallèle MPI), le vecteur solution est ensuite transmis, en entier, à tous les processus MPI. Le cycle peut ainsi continuer quelque soit la configuration suivante.
De plus, on peut superposer ou substituer à ce parallélisme MPI (qui fonctionne sur toutes les plate-formes), un autre niveau de parallélisme géré cette fois par le langage OpenMP . Celui-ci est cependant limité aux fractions de machine partageant physiquement la même mémoire (PC multi-cœurs ou nœuds de serveur de calcul).
Il ne permet pas de baisser les consommations mémoire mais par contre il accélère certains types de calcul et ce, avec une granularité plus faible que celle du MPI: il procure une meilleure accélération même si le flot de données/traitements n’est pas très important. C’est un schéma parallèle d’ordre «informatique» qui intervient principalement dans les opérations basiques d’algorithmes d’algèbre linéaire ( via par exemple la librairie BLAS).
Ce parallélisme peut être:
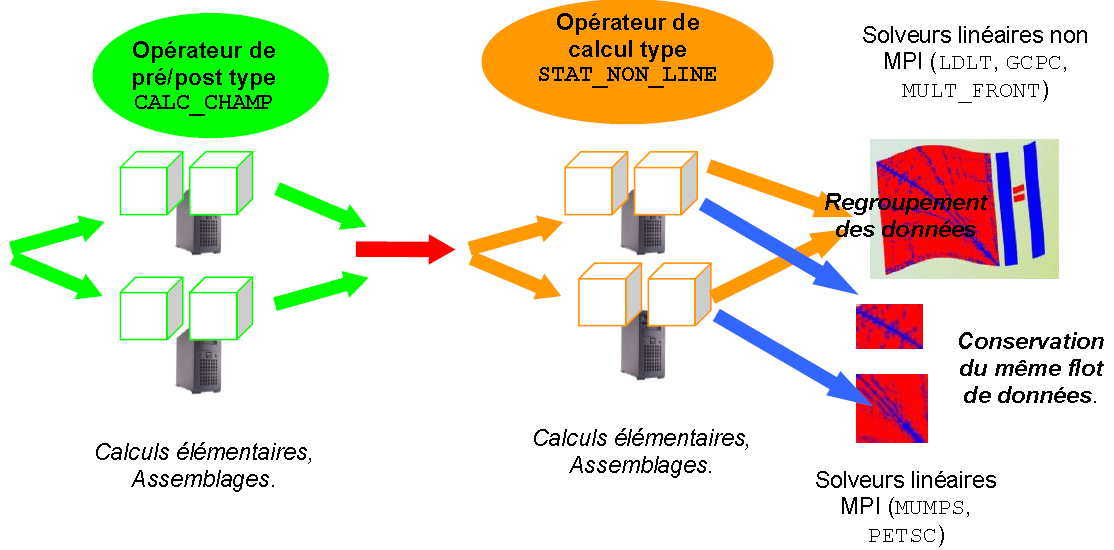
soit complémentaire du parallélisme MPI en accélérant les calculs au sein de chaque processus MPI (dans la partie résolution de système linéaire avec MUMPS, stratégie dite «2b/1c»).
soit se substituer au parallélisme MPI en accélérant la résolution de système linéaire avec MULT_FRONT (stratégie 2a).
Figure 2.3.1._ Organisation du schéma parallèle MPI
de construction et de résolution des systèmes linéaires.
Distribution de calculs modaux#
Lorsque la simulation ne peut pas se décomposer en calculs Aster indépendants, mais qu’elle reste dominée néanmoins par des calculs modaux généralisés (opérateurs INFO_MODE et CALC_MODES), on peut organiser un schéma parallèle spécifique.
Il est fondé sur la distribution de calculs modaux indépendants: chacun étant en charge d’une sous-bande fréquentielle. Ce schéma parallèle d’ordre purement «informatique» ne procure que des gains en temps.
Il peut toutefois cohabiter avec les schémas parallèles précédents:
chaînage avec le parallélisme MPI de construction des systèmes linéaires (dans par exemple CALC_MATR_ELEM,stratégie 1b+1d),
cumul avec le parallélisme MPI (voire OpenMP) dans les résolutions de systèmes linéaires (si utilisation de MUMPS,stratégie à 2 niveaux de parallélisme, 1d/2b voire trois, 1d/2b/1c).
Figure 2.4.1._ Organisation du schéma parallèle MPI de distribution des calculs modaux et de résolutions des systèmes linéaires associés.
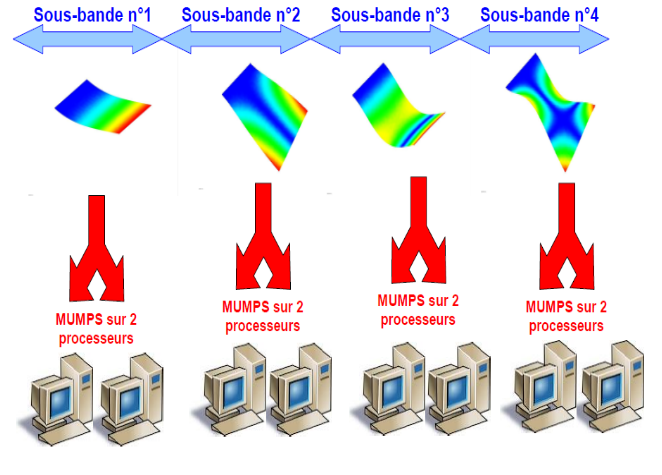
Remarque s :
Entre chaque commande, l’utilisateur peut même changer la répartition des mailles suivant les processeurs. Il lui suffit d’utiliser la commande MODI_MODELE. Ce mécanisme reste licite lorsqu’on enchaîne différents calculs (mode POURSUITE). La règle étant bien sûr que cette nouvelle répartition perdure, pour le modèle concerné, jusqu’à l’éventuel prochain MODI_MODELE et que cette répartition doit rester compatible avec le paramétrage parallèle du calcul (nombre de nœuds/processeurs…).
De toute façon, tout calcul parallèle Aster doit respecter les paradigmes suivant : en fin d’opérateur de calcul [6]_
, les bases globales de chaque processeur sont identiques [7]_ et le communicateur MPI courant est le communicateur standard ( MPI_COMM_WORLD ). Tous les autres éventuels sous-communicateurs MPI doivent être détruits.
Car on ne sait pas si l’opérateur qui suit dans le fichier de commandes a prévu un flot de données incomplet. Il faut donc organiser les communications idoines pour compléter les champs éventuellement incomplets.
Quelques conseils préalables#
On formule ici quelques conseilspour aider l’utilisateur à tirer parti des stratégies de calcul parallèle du code . Mais il faut bien être conscient, qu’avant tout chose, il faut d’abord optimiser et valider son calcul séquentiel en tenant compte des conseils qui fourmillent dans les documentations des commandes. Pour ce faire, on utilise, si possible, un maillage plus grossier et/ou on n’active que quelques pas de temps.
Le paramétrage par défaut et les affichages/alarmes du code proposent un fonctionnement équilibré et instrumenté . On liste ci-dessous et, de manière non exhaustive, plusieurs questions qu’il est intéressant de se poser lorsqu’on cherche à paralléliser son calcul. Bien sûr, certaines questions (et réponses) sont cumulatives et peuvent donc s’appliquer simultanément.
Préambule#
Il est intéressant de valider , au préalable, son calcul parallèle en comparant quelques itérations en mode séquentiel et en mode parallèle. Cette démarche permet aussi de calibrer les gains maximums atteignables (speed-up théoriques) et donc d’éviter de trop «gaspiller de processeurs». Ainsi, si on note \(f\) la portion parallèle du code (déterminée par exemple via un run séquentiel préalable), alors le speed-up théorique S p maximal accessible sur p processeurs se calcule suivant la formule d’Amdhal (cf. [R6.01.03] §2.4) :
\({S}_{p}=\frac{1}{1-f+\frac{f}{p}}\)
Par exemple, si on utilise le parallélisme MUMPS distribué par défaut (scénario 1b+2b) et que les étapes de construction/résolution de système linéaire représentent 90% du temps séquentiel ( f =0.90), le speed-up théorique est borné à la valeur \({\mathrm{S}}_{\infty}=\frac{1}{1-0.9+0.9/\infty }=10\) ! Et ce, quelque soit le nombre de processus MPI alloués.
Il est intéressant d’évaluer les principaux postes de consommation (temps/RAM) : en mécanique quasi-statique, ce sont généralement les étapes de calculs élémentaires/assemblages , de résolution de système linéaire et les algorithmes de contact-frottement . Mais leurs proportions dépendent beaucoup du cas de figure (caractéristiques du contact, taille du problème, complexité des lois matériaux…). Si les calculs élémentaires sont importants, il faut les paralléliser via la scénario 1b (scénario par défaut). Si, au contraire, les systèmes linéaires focalisent l’essentiel des coûts, les scénarios 2b ou 2c peuvent suffire. Par contre, si c’est le contact-frottement qui dimensionne le calcul, il faut chercher à optimiser son paramétrage et/ou paralléliser son solveur linéaire interne (cf. méthode GCP+MUMPS).
En dynamique, si on effectue un calcul par projection sur base modale, cela peut être cette dernière étape qui s’avère la plus coûteuse. Pour gagner du temps, on peut alors utiliser l’opérateur CALC_MODES avec l’option “BANDE” découpée en plusieurs sous-bandes, en séquentiel et, surtout, en parallèle (scénario 1d). Cet opérateur exhibe une distribution de tâches quasi-indépendantes qui conduit à de bons speedups.
Pour optimiser son calcul parallèle, il faut surveiller les éventuels déséquilibres de charge du flot de données (CPU et mémoire) et limiter les surcoûts dus aux déchargements mémoire (JEVEUX et MUMPS OOC) et aux archivages de champs . Sur le sujet, on pourra consulter la documentation [U1.03.03] «Indicateur de performance d’un calcul (temps/mémoire)». Elle indique la marche à suivre pour établir les bons diagnostics et elle propose des solutions.
Pour CALC_MODES avec l’option “BANDE” découpée en plusieurs sous-bandes, le respect du bon équilibrage de la charge est crucial pour l’efficacité du calcul parallèle : toutes les sous-bandes doivent comporter un nombre similaire de modes. On conseille donc de procéder en trois étapes:
Calibrage préalable de la zone spectrale par des appels à INFO_MODE(si possible en parallèle),
Examen des résultats,
Lancement en mode POURSUITE du calcul CALC_MODES, avec l’option “BANDE” découpée en plusieurs sous-bandes parallélisées.
Pour plus de détails on pourra consulter la documentation utilisateur de l’opérateur[U4.52.02].
Quelques chiffres empiriques#
On conseille d’allouer au moins 30 à 50.103 ddls par processus MPI (scénarios 1b, 2b ou 2c). Cette granularité peut descendre à quelques milliers de ddls par threads OpenMP tout en restant efficace (scénarios 1c ou 2a).
Un calcul thermo-mécanique standard bénéficie généralement, sur 32 processeurs, d’un gain de l’ordre de la dizaine en temps elapsed et d’un facteur 4 en pic mémoire RAM (par cœur).
Pour CALC_MODES, on conseille de décomposer son calcul en sous-bandes de quelques dizaines de modes (par exemple 60) et, ensuite, de prévoir 2, 4 voire 8 processus MUMPS par sous-bande. Il faut bien sûr composer avec le nombre de processeurs disponibles et le pic mémoire requis par le problème [8]_ .
On peut obtenir des gains d’un facteur 30, en temps elapsed, sur une centaine de processeurs. Les gains en mémoire sont plus modestes (quelques dizaines de pourcents).
L’étape de calibration modale via INFO_MODE ne coûte elle pratiquement rien en parallèle: quelques minutes, tout au plus, pour des problèmes de l’ordre du million d’inconnus, parallélisés sur une centaine de processeurs. Les gains en temps sont d’un facteur X70 sur une centaine de processeurs et jusqu’à x2 en pic mémoire RAM.
Calculs indépendants#
Lorsque la simulation que l’on souhaite effectuer se décompose naturellement (étude paramétrique, calcul de sensibilité…) ou, artificiellement (chaînage thermo-mécanique particulier…), en calculs similaires mais indépendants, on peut gagner beaucoup en temps calcul grâce au parallélisme numérique 1a.
Gain en mémoire RAM#
Lorsque le facteur mémoire dimensionnant concerne la résolution de systèmes linéaires (ce qui est souvent le cas), le cumul des parallélismes informatiques 1b (calculs élémentaires/assemblages) et numériques 2b (solveur linéaire distribué MUMPS) est tout indiqué [9]_ (voire 1b+2b/1c).
Une fois que l’on a distribué son calcul Code_Aster +MUMPS/PETSC sur suffisamment de processeurs, les consommations RAM de JEVEUX peuvent devenir prédominantes (par rapport à celles de MUMPS que l’on a étalées sur les processeurs). Pour rétablir la bonne hiérarchie (le solveur externe doit supporter le pic de consommation RAM !) il faut activer, en plus, l’option SOLVEUR/MATR_DISTRIBUEE [U4.50.01].
Pour résoudre des problèmes frontières de très grandes tailles (> 5M ddls), on peut aussi essayer les solveurs itératifs de PETSC (stratégie parallèle 2c ou 2b+2c).
Gain en temps#
Si l’essentiel des coûts concerne uniquement les résolutions de systèmes linéaires de petite taille (N<0.5M ddls) on peut se contenter d’utiliser les solveurs linéairesMUMPS en mode centralisé (stratégie 2b) ou MULT_FRONT (2a). Dès que la construction des systèmes devient non négligeable (>5%) ou que ceux-ci s’avèrent assez gros, il est primordial d’étendre le périmètre parallèle en activant la distribution des calculs élémentaires/assemblages (1b) et en passant à MUMPS distribué (valeur par défaut).
Sur des problèmes frontières de grandes tailles (N>3M ddls), une fois les bons paramètres numériques sélectionnés [10]_ (préconditionneur, relaxation… cf. [U4.50.01]), les solveurs itératifs parallèle (2c) peuvent procurer des gains en temps très appréciables par rapport aux solveurs directs génériques (2a/2b). Surtout si les résolutions sont relaxées [11]_ car, par la suite, elles sont corrigées par un processus englobant (algorithme de Newton de THER/STAT_NON_LINE…).
Pour les gros problèmes modaux (en taille de problème et/ou en nombre de modes), il faut bien sûr penser à utiliser CALC_MODES avec l’option “BANDE” découpée en plusieurs sous-bandes [12]_ .
Parallélismes informatiques#
Rampes de calculs indépendants#
Descriptif#
Utilisation: grand public via Astk .
Périmètre d’utilisation: calculs indépendants (paramétrique, étude de sensibilité…).
Nombre de cœurs conseillés: limite de la machine/gestionnaire batch .
Gain: en temps CPU.
Speed-up: proportionnel au nombre de cas indépendants.
Type de parallélisme: informatique via des scripts shell.
Scénario: 1a du §3. Cumul avec toutes les autres stratégies de parallélisme licite mais s’adressant à des utilisateurs avancés (hors périmètre d” Astk ).
Mise en œuvre#
L’outil Astkpermet d’effectuer toute une série de calculs similaires mais indépendants (en séquentiel et surtout en parallèle MPI). On peut utiliser une version officielle du code ou une surcharge privée préalablement construite. Les fichiers de commande explicitant les calculs sont construits dynamiquement à partir d’un fichier de commande «modèle» et d’un mécanisme de type «dictionnaire» : jeux de paramètres différents pour chaque étude (mot-clé VALE du fichier .distr), blocs de commandes Aster /Python variables (PRE/POST_CALCUL)…
Le lancement de ces rampes parallèles s’effectue avec les paramètres de soumission usuels d” Astk . On peut même reparamétrer la configuration matérielle du calcul (liste des nœuds, nombre de cœurs, mémoire RAM totale par nœud…) via un classique fichier .hostfile.
Pour plus d’informations sur la mise en œuvre de ce parallélisme, on pourra consulter les documentations [U1.04.00]/[U2.08.07].
Remarques :
Avant de lancer une telle rampe de calculs, il est préférable d’optimiser au préalable sur une étude type, les différents paramètres dimensionnants: gestion mémoire JEVEUX , aspects solveurs non linéaire/modaux/linéaire, mot-clé ARCHIVAGE, algorithme de contact-frottement… (cf. documentations [U1.03.03], [U4.50.01]…).
On peut facilement écrouler une machine en lançant trop de calculs vis-à-vis des ressources disponibles. Il est conseillé de procéder par étapes et de se renseigner quant aux possibilités d’utilisation de moyens de calculs partagés (classe batch, gros jobs prioritaires…).
Calculs élémentaires et assemblages#
Descriptif#
Utilisation: grand public via Astk .
Périmètre d’utilisation: calculs comportant des calculs élémentaires/assemblages coûteux (mécanique non linéaire). Activé par défaut dès que le nombre de processus MPI>1.
Nombre de cœurs conseillés: seul, entre 4 et 8. Chaîné avec le parallélisme distribué de MUMPS ou de PETSC (valeur par défaut), typiquement 16, 32 voire 64.
Gain: en temps voire en mémoire avec solveur linéaire MUMPS/PETSC (si MATR_DISTRIBUEE).
Speed-up: Gains variables suivant les cas (efficacité parallèle [13]_ >50%). Il faut une assez grosse granularité pour que ce parallélisme reste efficace : 30 ou 50.103 ddls par processus MPI.
Type de parallélisme: informatique via la langage MPI (mpi_nbcpu/mpi_nbnœud).
Scénario: 1b du §2. Nativement conçu pour se chaîner aux parallélismes numériques 2b ou 2c. Chaînage possible avec 1c, possible mais peu utile avec 1d ou 2a. Pas de cumul possible.
Mise en œuvre#
La mise en œuvre de ce schéma parallèle s’effectue de manière transparente pour l’utilisateur. Via Astk, elle s’initialise par défaut dès qu’on a sélectionné une version parallèle de Code_Aster (notée ***_mpi) ainsi qu’un nombre de processus MPI au moins égale à 2.
Ainsi sur le serveur centralisé Aster , il faut paramétrer les champs suivants dans le menu Options :
mpi_nbcpu=m , nombre de processus MPI alloués.
mpi_nbnœud=p , nombre de nœuds sur lesquels vont être distribués ces processus MPI.
Par exemple, sur la machine centralisée Aster5, les nœuds sont composés de 24 cœurs. Pour allouer 32 processus MPI à raison de 8 processus par nœud, il faut donc positionner mpi_nbcpu à 32 et mpi_nbnœud à 4.
On conseille, en général, de ne pas allouer tous les cœurs d’un nœud en MPI seul . Cela peut avoir pour effet de ralentir la simulation car, même si une partie des calculs s’en trouve accélérée du fait de sa distribution sur plus de cœurs, comme ceux-ci partagent certaines ressources mémoire, lesaccès aux donnéessont, eux, ralentis.
Pour utiliser plus efficacement et à 100% toutes les ressources allouées on conseille plutôt de panacherparallélisme MPI et OpenMP (cf. scénarios 1b+2b/1c ou 1b+2b/1c+2c).
Une fois ce nombre de processus MPI fixé, on peut lancer son calcul (en batch sur la machine centralisé) avec le même paramétrage qu’en séquentiel. Si ce schéma parallèle est chaîné avec le parallélisme numérique de MUMPS ou celui de de PETSC (ou les 2, cf. scénarios 2b et 2c), on peut réduire son pic mémoire RAM en activant l’option MATR_DISTRIBUEE.
Dès que plusieurs processus MPI sont activés , l’affectation du modèle dans le fichier de commandes Aster (opérateur AFFE_MODELE) distribue ses mailles entre les processeurs . Code_Aster étant un code éléments finis, c’est une distribution naturelle des données (et des tâches associées). Par la suite, les étapes Aster de calculs élémentaires et d’assemblages (matriciels et vectoriels) vont se baser sur cette distribution pour «tarir» les flots de données/traitements locaux à chaque processeur. Chaque processeur ne va effectuer que les calculs associés au groupe de maille dont il a la charge.
Cette répartition maille/processeur se décline de différentes manières et elle est paramétrable dans les opérateurs AFFE_MODELE[U4.41.01]/MODI_MODELE[U4.41.02] via les valeurs du mot-clé DISTRIBUTION/METHODE=:
“CENTRALISE”: Les mailles ne sont pas distribuées (comme en séquentiel). Chaque processeur connaît l’intégralité des mailles du modèle. Le parallélisme 1b n’est donc pas mis en œuvre. Ce mode d’utilisation est utile pour les tests de non-régression et pour certaines études où le parallélisme 1b rapporte peu voire est contre-productif (par ex. si on doit rassembler les données élémentaires pour nourrir un système linéaire non distribué et que les communications MPI requises sont trop lentes). Dans tous les cas de figure où les calculs élémentaires représentent une faible part du temps total (par ex. en élasticité linéaire), cette option peut être suffisante.
“GROUP_ELEM” /” MAIL_DISPERSE” /”MAIL_CONTIGU” /”SOUS_DOMAINE” (défaut) /”SOUS_DOM.OLD”: les mailles sont distribuées en se basant sur différents critères: par type, par distribution cyclique, par paquets de même taille ou suivant des stratégies sous-domaines.
Dans les deux derniers scénarios, la distribution s’effectue via les partitionneurs METIS (défaut) ou SCOTCH (cf. mot-clé PARTITIONNEUR). Ceux-ci doivent donc être installés et linkés à la versions Code_Aster utilisée. C’est évidemment fait par défaut sur la machine centralisée.
Remarques:
Entre chaque commande, l’utilisateur peut même changer la répartition des mailles suivant les processeurs. Il lui suffit d’utiliser la commande MODI_MODELE. Ce mécanisme reste licite lorsqu’on enchaîne différents calculs (mode POURSUITE). La règle étant bien sûr que cette nouvelle répartition doit rester compatible avec le paramétrage parallèle du calcul (nombre de nœuds/processeurs…).
Seul le parallélisme MPI permet d’accélérer cette étape du calcul. Or si on alloue trop de MPI, l’étape de résolution de système linéaire risque d’être ralentie ou d’être bloquée par manque de mémoire. Pour pallier à ce problème, on peut, avec le solveur linéaire MUMPS (cf. mot-clé SOLVEUR/REDUCTION_MPI), réduire le nombre de processus MPI utilisés uniquement dans MUMPS.
Les cœurs inoccupés se voient alors confier à l’autre niveau de parallélisme, l’OpenMP. La consommation mémoire globale du calcul s’en trouve ainsi réduite et ce, sans ralentir les résolutions de système linéaire. On accélère ainsi grandement l’étape de calcul élémentaire/assemblage sans impacter, ni le coût mémoire, ni celui en temps, de l’étape de résolution de système.
Structures de données distribuées#
La distribution des données qu’implique ce type de parallélisme numérique ne diminue pas forcément les consommations mémoire JEVEUX . Par soucis de lisibilité/maintenabilité, les objets Code_Aster usuels sont initialisés avec la même taille qu’en séquentiel. Chaque processus MPI se «contente» juste de les remplir partiellement avec les données produites par les mailles dont a la charge le processeur. A charge pour le solveur linéaire parallèle utilisé dans la suite de l’opérateur (MUMPS, PETSC ou les 2) d’assembler ces données incomplètes et distribuées. On ne retaille donc généralement pas ces structures de données, elles comportent beaucoup de valeurs nulles.
Cette stratégie n’est tenable que tant que les objets JEVEUX principaux impliqués dans les calculs élémentaires/assemblages (CHAM_ELEM, RESU_ELEM, MATR_ASSE et CHAM_NO) ne dimensionnent pas les contraintes mémoire du calcul (cf. §5 de [U1.03.03]).
Normalement leur occupation mémoire est négligeable comparée à celle du solveur linéaire. Mais lorsque ce dernier (par ex. MUMPS) est lui aussi Out-Of-Core [14]_ et parallélisé en MPI (avec la répartition des données entre processeurs que cela implique), cette hiérarchie n’est plus forcément respectée. D’où l’introduction d” une option (mot-clé MATR_DISTRIBUEE cf. [U4.50.01]) permettant de véritablement retailler, au plus juste, le bloc de matrice Asterpropre à chaque processus MPI.
Remarque:
En mode distribué, chaque processus MPI ne manipule que des matrices incomplètes (retaillées ou non). Par contre, afin d’éviter de nombreuses communications MPI (lors de l’évaluation des critères d’arrêt, calculs de résidus…), ce scénario n’a pas été retenu pour les vecteurs seconds membres. Leurs constructions sont bien parallélisées, mais, à l’issue de l’assemblage, les contributions partielles de chaque processus sont rassemblées. Ainsi, tout processus MPI connaît entièrement les vecteurs (CHAM_NO) impliqués dans le calcul.
Distribution des calculs d’algèbre linéaire basiques#
Descriptif#
Utilisation: grand public viaAstk.
Périmètre d’utilisation: calculs comportant des résolutions de systèmes linéaires via MUMPS (usage solveur direct ou préconditionneur de PETSC/GCPC).
Nombre de cœurs conseillés: sur Aster5, entre 2 et 12. Pas plus que de cœurs physiques partageant la même mémoire physique.
Gain: en temps elapsed (mais augmentation du temps CPU).
Speed-up: Gains variables suivant les cas (efficacité parallèle [15]_ >50%). Une granularité faible suffit pour que ce parallélisme reste efficace : 10.103 ddls par threads OpenMP.
Type de parallélisme: informatique via le langage OpenMP (ncpus).
Scénario: 1c du §2. Une utilisation classique consiste à tirer parti d’un parallélisme hybride MPI+OpenMP pour accentuer les performances de MUMPS et de tirer partie à 100% des ressources machine (2b/1c ou (2b/1c)+2c).
Cumul contre-productif avec 2a, utile avec 2b, possible mais peu utile avec 2c (seul) ou 1d.
Mise en œuvre#
La mise en œuvre de ce schéma parallèle s’effectue de manière transparente pour l’utilisateur. Via Astk, elle s’initialise par défaut dès qu’on a sélectionné une version parallèle de Code_Aster (notée ***_mpi) ainsi qu’un nombre de threads OpenMP au moins égale à 2.
Ainsi sur le serveur centralisé Aster , il faut paramétrer les champs suivants dans le menu Options :
ncpus=k , nombre de threads OpenMP alloués (par processus MPI si mpi_nbcpu>1).
Ce schéma parallèle est généralement utilisé en conjonction du parallélisme MPI de MUMPS. Car on conseille, en général, de ne pas allouer tous les cœurs d’un nœud en MPI seul . Cela peut avoir pour effet de ralentir la simulation car, même si une partie des calculs s’en trouve accélérée du fait de sa distribution sur plus de cœurs, comme ceux-ci partagent certaines ressources mémoire, les accès aux données sont, eux, ralentis. Pour utiliser plus efficacement et à 100% toutes les ressources allouées on conseille plutôt de panacher parallélisme MPI et OpenMP (cf. scénarios 2b/1c ou (2b/1c)+2c).
Dans ce type de parallélisme hybride (MPI/OpenMP), l’outil Astk[U1.04.00] complète automatiquement le nombre de threads en fonction des ressources machines, dès que le champ ncpus est laissé vide.
Par exemple, sur la machine centralisée Aster5 , les nœuds sont composés de 24 cœurs. Si on souhaite organiser un parallélisme hybride 12 MPI x 4 OpenMP, il suffit de positionner mpi_nbcpu à 12, mpi_nbnœud à 2 et ncpus=<vide> (ou 4 explicitement).
Remarque:
La mise en œuvre de ce parallélisme dépend du contexte informatique (matériel, logiciel) et des librairies d’algèbre linéaire utilisées. Sur la machine centralisée Aster, on utilise les BLAS threadées MKL.
Calculs modaux d’INFO_MODE/CALC_MODES#
Descriptif#
Utilisation: grand public viaAstk.
Périmètre d’utilisation: calculs comportant de coûteuses recherches de modes propres.
Nombre de cœurs conseillés: plusieurs dizaines (par exemple, nombre de sous-bandes fréquentielles x 2, 4 ou 8).
Gain : en temps elapsed v oire en mémoire RAM (grâce au deuxième niveau de parallélisme).
Speedup: Gains variables suivant les cas: efficacité de l’ordre de 70% sur le premier niveau de parallélisme (sur les sous-bandes fréquentielles) complété par le parallélisme éventuel du second niveau (si SOLVEUR=MUMPS, efficacité complémentaire de l’ordre de 20%).
Type de parallélisme: informatique via le langage MPI (mpi_nbcpu/mpi_nbnœud).
Scénario: 1d du §2. Nativement conçu pour se coupler au parallélisme 2b (voire 2b/1c).
Chaînage possible mais peu utile avec 1b. Cumul possible mais peu utile avec 2a et impossible avec 2c (hors périmètre).
Mise en œuvre#
L’usage de CALC_MODES avec l’option “BANDE” découpée en plusieurs sous-bandes est à privilégier lorsqu’on traite des problèmes modaux de tailles moyennes ou grandes (>0.5M ddls) et/ou que l’on cherche une bonne partie de leurs spectres (> 50 modes).
On découpe alors le calcul en plusieurs sous-bandes fréquentielles. Sur chacune de ces sous-bandes, un solveur modal effectue la recherche de modes associée. Pour ce faire, ce solveur modal utilise intensivement un solveur linéaire.
Ces deux briques de calcul (solveur modal et solveur linéaire) sont les étapes dimensionnantes du calcul en terme de consommation mémoire et temps. C’est sur elles qu’il faut mettre l’accent si on veut réduire significativement les coûts calcul de cet opérateur (cf. figures 1.3a/b).
Or, l’organisation du calcul modal sur des sous-bandes distinctes offre ici un cadre idéal de parallélisme: distribution de gros calculs presque indépendants . Son parallélisme permet de gagner beaucoup en temps mais au prix d’un surcoût en mémoire [16]_ .
Si on dispose d’un nombre de processeurs suffisant (> au nombre de sous-bandes non vides), on peut alors enclencher un deuxième niveau de parallélisme viale solveur linéaire (si on a choisit METHODE=”MUMPS”). Celui-ci permettra de continuer à gagner en temps mais surtout, il permettra de compenser le surcoût mémoire du premier niveau voire de diminuer notablement le pic mémoire séquentiel.
Pour un usage optimal de CALC_MODES avec l’option “BANDE” découpée en plusieurs sous-bandes parallélisées, il est donc conseillé de:
Construire des sous-bandes de calcul relativement équilibrées . Pour ce faire, on peut donc, au préalable, calibrer le spectre étudié via un appel à INFO_MODE [U4.52.01] (si possible en parallèle). Puis lancer le calcul CALC_MODES avec l’option “BANDE” découpée en plusieurs sous-bandes parallélisées en fonction du nombre de sous-bandes choisies et du nombre de processeurs disponibles.
De prendre des sous-bandesentre 50 et 100modes .
Sélectionnerun nombre de processeurs qui est un multiple du nombre de sous-bandes (non vides). Ainsi, on réduit les déséquilibrages de charges qui nuisent aux performances.
Pour plus de détails on pourra consulter la documentation utilisateur de l’opérateur[U4.52.02].
Calculs MISS3D via CALC_MISS#
Descriptif#
Utilisation: grand public viaAstk.
Périmètre d’utilisation: calculs comportant des calculs MISS3D.
Nombre de cœurs conseillés: avec option FICHIER_TEMPS, plusieurs dizaines (par exemple, nombre de fréquences x4 ou X8); autre option, jusqu’à 24 cœurs.
Gain : en temps elapsed .
Speed-up: Gains variables suivant les cas: efficacité de l’ordre de 100% sur le premier niveau de parallélisme (en MPI sur les fréquences si FICHIER_TEMPS) complété par le parallélisme éventuel du second niveau (en OpenMP).
Type de parallélisme: informatique via le langage MPI (mpi_nbcpu/mpi_nbnœud) si FICHIER_TEMPS, complété éventuellement par un parallélisme OpenMP (toutes les autres options de CALC_MISS, ncpus) .
Scénario: 1e du §2.
Mise en œuvre#
Si les calculs CALC_MISS sont indépendants (option FICHIER_TEMPS), on peut déjà les distribuer sur plusieurs nœuds, en réservant plusieurs processus MPI par nœud. Chacun va traiter une série de fréquences. On ne prendra pas tous les c œ urs d’un nœud afin de laisser assez de mémoire RAM au calcul MISS3D.
Ensuite, afin d’utiliser au mieux ces cœurs inoccupés, il est alors conseillé de rajouter un deuxième niveau de parallélisme en activant des threads OpenMP au sein de chaque processus MPI MISS3D (cf. figures 1.4a/b).
Si les calculs ces calculs CALC_MISS ne sont, par contre, pas indépendants (autre option que FICHIER_TEMPS), on n’utilisera qu’un nœud du cluster et on n’activera que le second niveau de parallélisme via OpenMP.
Pour plus de détails on pourra consulter la documentation utilisateur de l’opérateur[U2.06.07].
Autres distributions de calculs#
Descriptif#
Utilisation: grand public viaAstk.
Périmètre d’utilisation: calculs comportant du contact-frottement, des appels à REST_SPEC_TEMP[U4.63.34], CALC_CHAMP[U4.81.04] (options REAC/FORC_NODA et ***_NOEU)…
Nombre de cœurs conseillés: pas de paramétrage spécifique (mode parallèle par défaut), garder le même paramétrage parallèle MPI que pour le parallélisme de MUMPS: au moins 50000 ou 100000 ddls par processus MPI.
Sauf pour le «parallélisme en temps» de CALC_CHAMP, qui lui est efficace dès qu’on réserve au moins une douzaine pas de temps par processus MPI. Du moment que le calcul unitaire d’un seul pas de temps s’avère assez coûteux.
Gain : en temps elapsed .
Speed-up: Gain assez important si taille ci-dessus respectée: efficacité >50%.
Type de parallélisme: informatique via le langage MPI (mpi_nbcpu/mpi_nbnœud).
Scénario: 1f du §2.
Mise en œuvre#
Mise en œuvre identique à celle de MUMPS (cf. §5.5.2), sauf pour CALC_CHAMP[U4.81.04], pour lequel il faut activer un mot-clé spécifique (PARALLELISME_TEMPS=’OUI’).
Parallélismes numériques#
Solveur direct MULT_FRONT#
Descriptif#
Utilisation: grand public via Astk .
Périmètre d’utilisation: calculs comportant des résolutions de systèmes linéaires coûteuses (en général STAT/DYNA_NON_LINE, MECA_STATIQUE…).
Nombre de cœurs conseillés: 2 ou 4.
Gain : en temps elapsed.
Speedup: Gains variables suivant les cas (efficacité parallèle≈50%). Il faut une bonne granularité pour que ce parallélisme reste efficace : au moins 50.103 ddls par cœur.
Type de parallélisme: numérique via le langage OpenMP (ncpus).
Scénario: 2a du §2. Chaînage possible mais peu utile avec 1b, 2b ou 2c. Cumul contre-productif avec 1c, possible mais peu utile avec 1d.
Mise en œuvre#
Cette méthode multifrontale développée en interne (cf. [R6.02.02] ou [U4.50.01] §3.5) est utilisée via la mot-clé SOLVEUR/METHODE=”MULT_FRONT”. C’est le solveur linéaire (historique et auto-portant) préconisé par défaut en séquentiel sur les modèles de taille petite ou moyenne (<0.5M ddls).
La mise en œuvre de ce parallélisme s’effectue de manière transparente pour l’utilisateur. Elle s’initialise par défaut dès qu’on lance un calcul via Astk (menu Options) utilisant plusieurs threads OpenMP.
Ainsi sur le serveur centralisé Aster , il faut paramétrer le champs suivants:
ncpus=n, nombre de threads OpenMP alloués.
Une fois ce nombre de threads fixé on peut lancer son calcul (en batch sur la machine centralisé) avec le même paramétrage qu’en séquentiel. On peut bien sûr baisser les spécifications en temps du calcul.
Package MUMPS#
Descriptif#
Utilisation: grand public via Astk .
Périmètre d’utilisation: calculs comportant des résolutions de systèmes linéaires coûteuses (en général STAT/DYNA_NON_LINE, MECA_STATIQUE…).
Nombre de cœurs conseillés: chaîné avec le parallélisme distribué des calculs élémentaires/assemblages typiquement 16, 32 voire 64.
Gain: en temps CPU et en mémoire RAM.
Speedup: Gains variables suivant les cas (efficacité parallèle≈30%). Il faut une granularité moyenne pour que ce parallélisme reste efficace : entre 30 et 50.103 ddls par processus MPI.
Type de parallélisme: numérique via le langage MPI.
Scénario: 2b du §3. Nativement conçu pour se chaîner aux parallélismes 1b ou 2c. Chaînage possible mais peu utile avec 2a. Couplage très utile avec 1c ou 1d (voire les 2).
Mise en œuvre#
Cette méthode multifrontale s’appuie sur le produit externe MUMPS (cf. [R6.02.03] ou [U4.50.01] §3.7) est utilisée soit en tant que solveur direct (mot-clé SOLVEUR/METHODE=”MUMPS”), soit en tant que préconditionneur des solveurs itératifs PETSC ou GCPC (mot-clé SOLVEUR/PRE_COND=”LDLT_SP”).
C’est le package HPC conseillé pour exploiter pleinement les gains CPU/RAM que peut procurer le parallélisme . Ce type de parallélisme est performant (surtout lorsqu’il est chaîné avec 1b et couplé avec 1c) tout en restant générique, robuste et grand public.
La mise en œuvre de ce schéma parallèle s’effectue de manière transparente pour l’utilisateur. Via Astk, elle s’initialise par défaut dès qu’on a sélectionné une version parallèle de Code_Aster (notée ***_mpi) ainsi qu’un nombre de processus MPI au moins égale à 2.
Ainsi sur le serveur centralisé Aster , il faut paramétrer les champs suivants dans le menu Options :
mpi_nbcpu=m , nombre de processus MPI alloués.
mpi_nbnœud=p , nombre de nœuds sur lesquels vont être distribués ces processus MPI.
Par exemple, sur la machine centralisée Aster5, les nœuds sont composés de 24 cœurs. Pour allouer 32 processus MPI à raison de 8 processus par nœud, il faut donc positionner mpi_nbcpu à 32 et mpi_nbnœud à 4.
On conseille, en général, de ne pas allouer tous les cœurs d’un nœud en MPI seul . Cela peut avoir pour effet de ralentir la simulation car, même si une partie des calculs s’en trouve accélérée du fait de sa distribution sur plus de cœurs, comme ceux-ci partagent certaines ressources mémoire, lesaccès aux donnéessont, eux, ralentis.
Pour utiliser plus efficacement et à 100% toutes les ressources allouées on conseille plutôt de panacherparallélisme MPI et OpenMP (cf. scénarios 1b+2b/1c ou 1b+2b/1c+2c).
Idéalement, ce solveur linéaire HPC doit être utilisé en mode parallèle distribué (DISTRIBUTION/METHODE=”GROUP_ELEM”/”MAIL_DISPERSE”/”MAIL_CONTIGU”/”SOUS_DOMAINE”/”SOUS_DOM.OLD”). C’est-à-dire qu’il faut avoir initié en amont de ce solveur linéaire, au sein des procédures de calculs élémentaires/assemblages, des flots de données/traitements distribués (scénario parallèle 1b). MUMPS accepte en entrée ces données incomplètes et il les rassemble en interne. On ne perd pas ainsi de temps (comme c’est le cas pour les autres solveurs linéaires) à compléter les données issues de chaque processeur. Ce mode de fonctionnement est activé par défaut dans les commandes AFFE/MODI_MODELE (cf. §4.2).
En mode centralisé (CENTRALISE), la phase amont de construction des systèmes linéaires n’est pas parallélisée (chaque processeur procède comme en séquentiel). MUMPS ne tient alors compte que des données issues du processeur maître.
Dans le premier cas, le code est parallèle de la construction du système linéaire jusqu’à sa résolution (chaînage des parallélismes 1b+2b), dans le second cas, on n’exploite le parallélisme MPI que sur la partie résolution (parallélisme 2b).
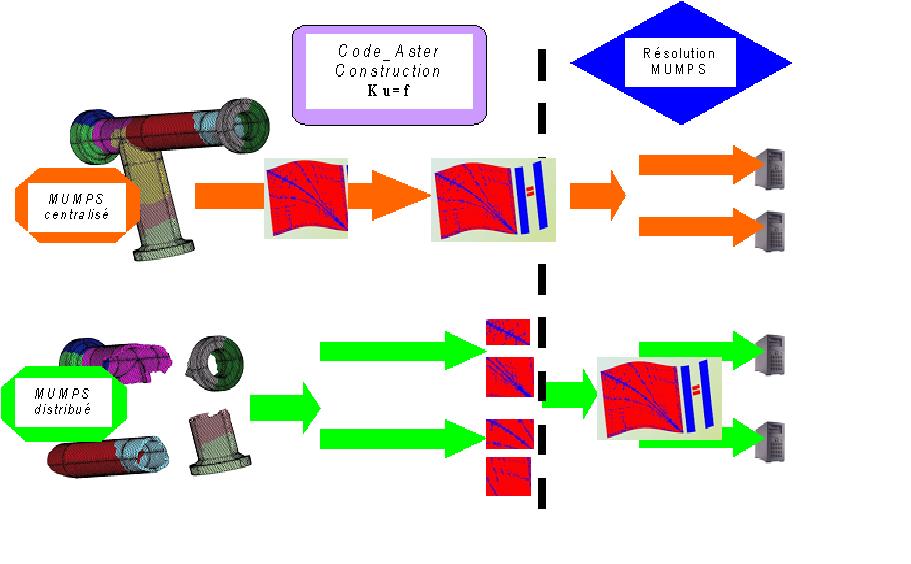
Figure 5.2.1._ Flots de données/traitements parallèles du couplage Code_Aster+ MUMPS suivant le mode d’utilisation:
centralisé ou distribué.
Remarque:
Lorsque la part du calcul consacrée à la construction du système linéaire est faible (<5%), les deux modes d’utilisation (centralisé ou distribué) affichent des gains en temps similaires. Par contre, seule l’approche distribuée procure, en plus, des gains sur les consommations RAM.
Solveur itératif PETSC#
Descriptif#
Utilisation: grand public via Astk .
Périmètre d’utilisation: calculs comportant des résolutions de systèmes linéaires coûteuses (en général STAT/DYNA_NON_LINE, MECA_STATIQUE…). Plutôt des problèmes non linéaires de grandes tailles.
Nombre de cœurs conseillés: chaîné avec le parallélisme distribué des calculs élémentaires/assemblages (1b), voire celui du préconditionneur MUMPS (2b), typiquement 16, 32 voire 64.
Gain: en temps CPU et en mémoire RAM (suivant les préconditionneurs).
Speedup: gains variables suivant les cas (efficacité parallèle>50%). Il faut une granularité moyenne pour que ce parallélisme reste efficace : 50.103 ddls par processus MPI.
Type de parallélisme: numérique via le langage MPI.
Scénario: 2c du §3. Nativement conçu pour se chaîner aux parallélismes 1b ou 2b; chaînage possible mais peu utile avec 2a. Cumul possible mais peu utile avec 1c, hors-périmètre avec 1d.
Mise en œuvre#
Cette bibliothèque de solveurs itératifs (cf. [R6.01.02] ou [U4.50.01] §3.9) est utilisée via la mot-clé SOLVEUR/METHODE=”PETSC”. Ce type de solveur linéaire est conseillé pour traiter, soit des problèmes frontières de très grande taille (>5M ddls), soit en non linéaire, pour tirer pleinement partie de la mutualisation du préconditionneur entre différents pas de Newton .
La mise en œuvre de ce parallélisme s’effectue comme pour le package MUMPS (cf. §5.2).
Remarque:
Contrairement aux solveurs parallèles directs (MUMPS, MULT_FRONT), les itératifs ne sont pas universels (ils ne peuvent pas être utiliser en modal) et toujours robustes. Ils peuvent être très compétitifs (en temps et surtout en mémoire), mais il faut trouver le point de fonctionnement (algorithme, préconditionneur…) adapté au problème.Toutefois, sur ce dernier point, l’usage généralisé (et paramétré par défaut) de MUMPSsimple précision comme préconditionneur (PRE_COND=”LDLT_SP”) a considérablement amélioré les choses.